Storicamente, il quadro clinico della Sindrome di Pendred, fu descritto nel 1896 dal Dr. Vaughan Pendred, medico inglese, il quale, per la prima volta, segnalò il caso di una famiglia irlandese con due sorelle (di dieci figli) affette da sordomutismo congenito e gozzo tirodeo, non correlabili in alcun modo a fattori acquisiti o ambientali.
Nonostante il Dr. Pendred intuì lereditarietà della sindrome, solo nel 1956 fu dimostrata la modalità di trasmissione autosomica recessiva del fenotipo.
Due anni più tardi, si attribuì il gozzo tiroideo a difetti di organificazione dello iodio a livello tiroideo con conseguente deficit di sintesi di tiroxina.
Nel 1967 e nel 1978 vennero descritte le tipiche malformazioni dellorecchio interno della sindrome di Pemdred ossia la malformazione tipo Mondini (con appiattimento della coclea e sviluppo del solo giro basale) e la dilatazione dellacquedotto vestibolare, questultima di gran lunga più comune e frequente. |
 |
| |
DR. VAUGHAN PENDRED (1869-1945)
Unità di Pediatria genetica e Medicina Fetale di Londra - Dipartimento di genetica Univ. Di Leicester |
Dal punto di vista epidemiologico, la sindrome di Pendred ha unincidenza di circa 9 affetti ogni 100.000 nuovi nati.
La sindrome, non curabile, non compromette la prognosi quoad vitam del paziente, né tanto meno le sue capacità riproduttive. Si suppone, ad oggi, che dall1 all8% delle ipoacusie congenite sia attribuibile alla Sindrome di Pendred.
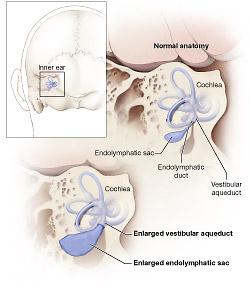 |
La sindrome di Pendred è una malattia genetica a carattere autosomico recessivo che clinicamente si manifesta, generalmente prima delladolescenza, con sordità congenita bilaterale neurosensoriale, a volte vertigini, EVA (Enlarged Vestibular Aqueduct), disfunzione tiroidea variabile sino al gozzo ipotiroideo e disordini della funzionalità renale (di solito sub-clinici), diagnosticabili solo con esami specifici.
La perdita delludito è presente sin dalla nascita, rendendo lapprendimento del linguaggio un problema di comune riscontro nellinfanzia di questi pazienti.
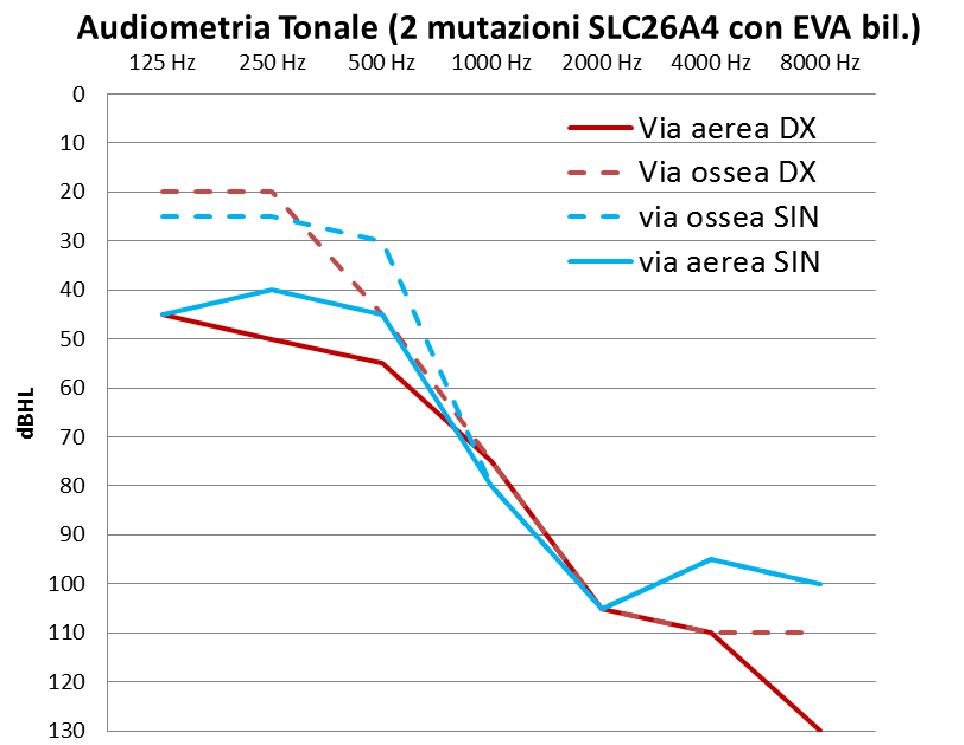
Lipoacusia, anche se prevalentemente neurosensoriale, può presentare delle componenti trasmissive, avere uninsorgenza più tardiva, essere fluttuante e progressiva; solitamente è di grado profondo con localizzazione prevalente sulle alte frequenze determinando un audiogramma in discesa sulle frequenze acute. |
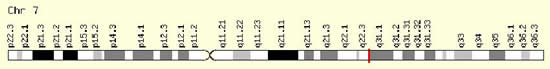
Etiogeneticamente, la Sindrome di Pendred è legata alla mutazione del gene SLC26A4 localizzato sul braccio lungo del cromosoma 7.
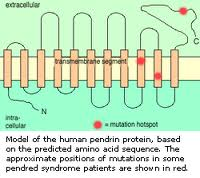
Il gene SLC26A4 (SoLute Carrier; secondo altre fonti SLC viene da Solute Linked Carrier) costituito da 21 esoni, porta quindi alla formazione di una proteina, detta Pendrina, di 780 ami-noacidi, localizzata nella membrana cellulare (detta pertanto transmembrana) con la specifica funzione di trasportare soluti con carica negativa e nella fattispecie iodio (I-), cloro (Cl-) e bicarbonato (HCO3-); è detta anche trasportatore cloro/iodio sodio-indipendente e fa parte della famiglia 26 che ha il nome più generico di scambiatori anio-nici multifunzione.
Il gene, localizzato sul braccio lungo del cromosoma 7 (7q21-34), pur essendo presente nel DNA di tutte le cellule, non è espresso ovunque, cioè la pendrina non si trova in tutte le cellule dellorganismo.
La Pendrina è espressa infatti nella tiroide, nellorecchio interno e nel rene.
Un suo difetto funzionale provoca compromissione nel trasporto di iodio, disfunzione tiroidea variabile e spesso gozzo.
Il gozzo e lipotiroidismo si manifestano generalmente a partire dalladolescenza ma possono presentarsi sin dalla nascita oppure apparire tardivamente. |
 |
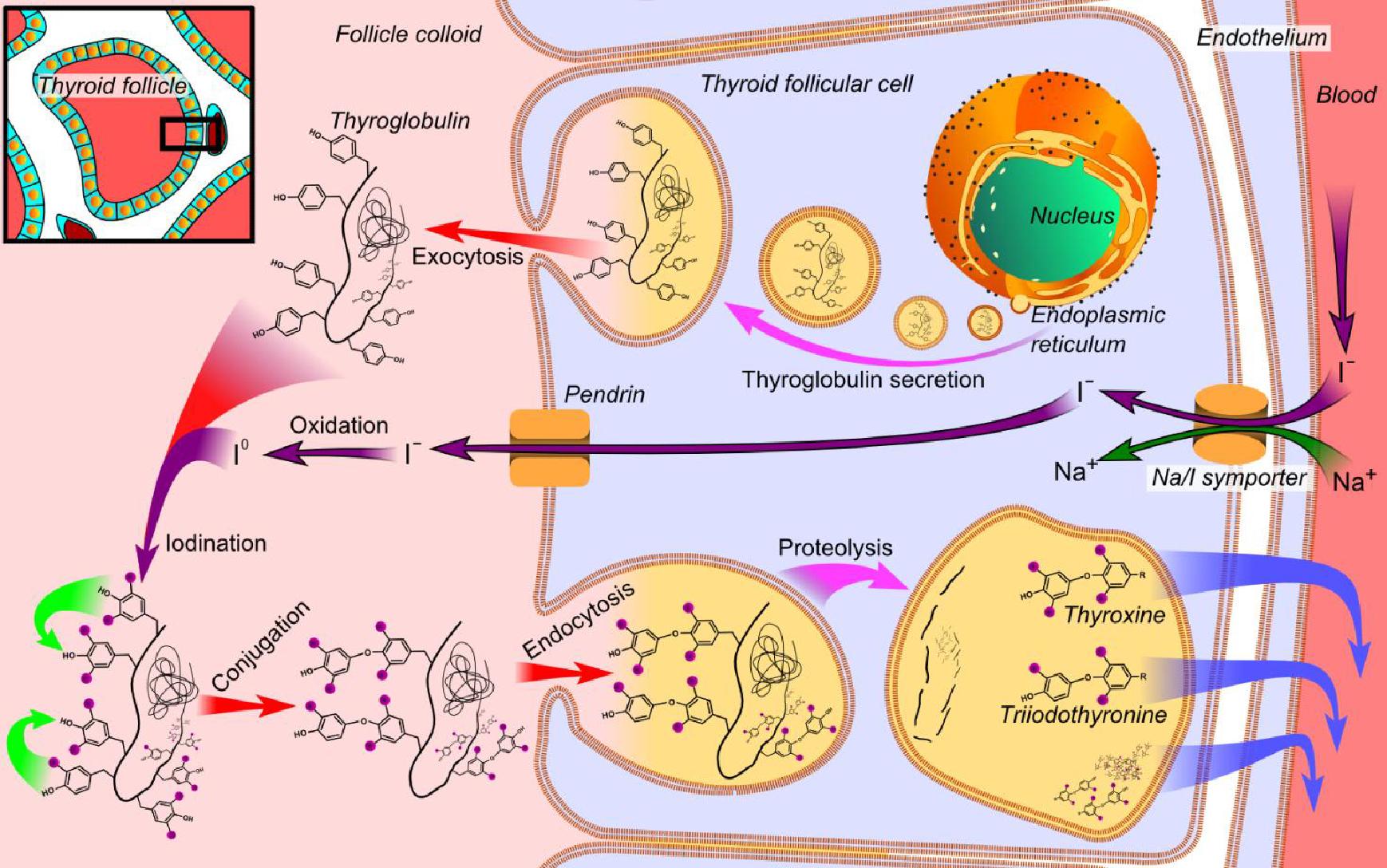
Il gene potrebbe però essere responsabile anche di un tipo sordità isolata con assenza di gozzo e di ipotiroidismo.
Infatti alcuni classificano la condizione di Pendred in due sottotipi: quella sindromica, associata a disfunzione tiroidea e gozzo e quella non sindromica in cui vi è la dilatazione dellacquedotto vestibolare senza ipertrofia/disfunzione ghiandolare tiroidea.
Nella Sindrome di Pendred, a livello dellorecchio interno, si determina, con meccanismo diverso, disfunzione/danno delle cellule sensoriali della coclea, dove la pendrina è presente sia nelle membrane delle cellule di Deiters, di Claudius, del legamento spirale, del ganglio spirale e del solco esterno ma anche anche nel vestibolo e nel sacco endolinfatico.
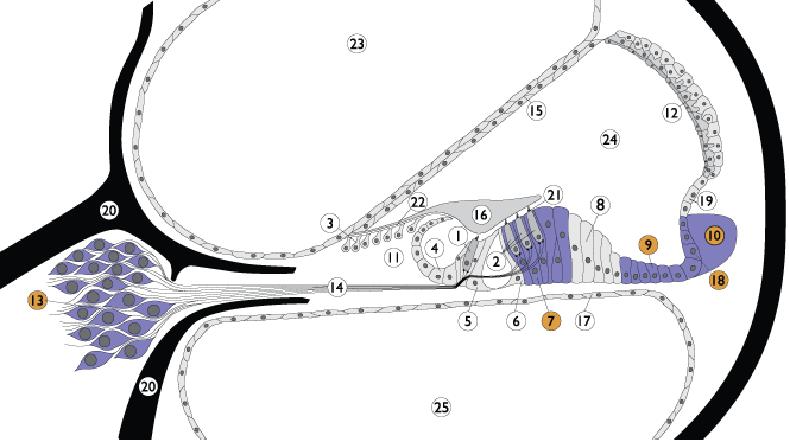
Nella coclea la pendrina è presente nella membrane cellulari del ganglio spirale, del legamento spirale, delle cellule di Claudius, di Dei-ters e del solco esterno.
Legenda: 1. Inner hair cell; 2. Outer hair cell; 3. Interdental cells; 4. Inner sulcus cells; 5. Inner pillar cells; 6. Outer pillar cells; 7. Deiters cells; 8. Hensen cells; 9. Claudius cells; 10. Spiral ligament; 11. Spiral limbus; 12. Stria vascularis; 13. Spiral ganglion; 14. Auditory nerve; 15. Reissners membrane; 16. Tectorial membrane; 17. Basilar membrane; 18. External sulcus cells; 19. Spiral promi-nence; 20. Bony spiral lamina; 21. Reticular lamina; 22. Between IDC and TM; 23. Scala vestibuli; 24. Scala media; 25. Scala tympani
Nellorecchio interno, la funzione della pendrina è differente rispetto alla ghiandola tiroidea: prima di tutto, non trasporta iodio, ma attua uno scambio tra ione cloro (Cl-) e ione bicarbonato (HCO3-); secondo, questo scambio non porta alla formazione di ormoni, ma semplicemente mantiene/regola lequilibrio elettrolitico dei liquidi endolinfatici.
Una funzione simile ha la pendrina nel rene, ma come spesso accade tra questi due organi (che presentano numerose analogie in termini di espressione e funzione proteica) la direzione dello scambio è diametralmente opposta. Ciò ovviamente in relazione alle diverse necessità e funzioni dei due organi (Fonte: Life-threatening metabolic alkalosis in Pendred syndrome Kandasamy et al., 2011).

Diverse funzioni della pendrina nei principali organi dove è espressa; notare che cambiano le molecole interessate e la direzione degli scambi, anche se la localizzazione è sempre apicale ed endoluminale.
Immagine tratta da Life-threatening metabolic alkalosis in Pendred syn-drome Kandasamy et al., 2011).
La diagnosi in età prenatale è effettuabile mediante un esame sul DNA di cellule fetali o mediante lanalisi dei villi coriali.
Successivamente con:
- Valutazione audiologica con esami (OAEs, ABR, Impedenzometria, COR, ASSR, audiometria tonale e vocale, quando possibile);
- TAC e/o RMN dellorecchio;
- Studio della funzionalità tiroidea ed esame ecografico ghiandolare;
- Studio della funzionalità renale ed esame ecografico app urinario (anche se raramente si hanno anomalie);
- Test al perclorato di K (Pertiroid);
Indagini genetiche (sequenziamento completo, incluse le regioni non tradotte del gene SLC26A4).
Dal punto di vista terapeutico non esistono trattamenti specifici per questo tipo di malattia.
Non è possibile infatti, ad oggi, guarire dalle mutazioni del gene SLC26A4, dallEVA o dalla Sindrome di Pendred, anche se la ricerca sta compiendo passi da gigante nellambito della terapia genica e delle cellule staminali.
È possibile però contrastare in maniera efficace e tempestiva i sintomi, tanto da permettere al paziente una vita del tutto normale. Per quanto riguarda lipoacusia, ovviamente, il trattamento è rappresentato dalla riabilitazione con protesi acustica o con impianto cocleare. Possono essere dausilio anche supporti del linguaggio Per quanto riguarda i disordini della tiroide, qualora dovessero essere presenti, previa valutazione specialistica dellendocrinologo, è solitamente sufficiente una terapia farmacologica cronica sostitutiva con levo-tiroxina nei casi di gozzo ipotiroideo.
BIOGRAFIA:
- Syndrome of the month: Pendred Syndrome, J.Med.Genet. 1996;33:1037-1040
- http://www.nidcd.nih.gov/health/hearing/pages/pendred.aspx, ( Pendred Syndrome)
- http://www.ncbi.nlm.nih.gov/books/NBK1467/, (Pendred Syndrome / DFNB4)
- http://www.medicinenet.com/pendred_syndrome/article.htm, (Pendred Syndrome)
- Significance of unilateral enlarged vestibular aqueduct, Greinwald J, Dealarcon A, Cohen A, Uwiera T, Zhang K, Benton C, Halstead M, Meinzen-Derr J. Laryngoscope. 2013 Feb 9. doi: 10.1002/lary.
- Molecular analysis of SLC26A4 gene in patients with nonsyndromic hearing loss and EVA: identification of two novel mutations in Brazilian patients, de Moraes VC, dos Santos NZ, Ramos PZ, Svidnicki MC, Castilho AM, Sartorato EL. Int J Pediatr Otorhinolaryngol. 2013 Mar;77(3):410-3. doi: 10.1016/j.ijporl.2012.11.042. Epub 2012 Dec 27.
- Pendrin: the thyrocyte apical membrane iodide transporter? Twyffels L, Massart C, Golstein PE, Raspe E, Van Sande J, Dumont JE, Beauwens R, Kruys V. Cell Physiol Biochem. 2011;28(3):491-6. doi: 10.1159/000335110.
- Pendred syndrome. Glaser B. Pediatr Endocrinol Rev. 2003 Dec;1 Suppl 2:199-204; discussion 204. Review
- The Pendred syndrome gene encodes a chloride-iodide transport protein. Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. Nat Genet. 1999 Apr;21(4):440-3.
- Related citations
- Goitrous congenital hypothyroidism and hearing impairment associated with mutations in the TPO and SLC26A4/PDS genes. Pfarr N, Borck G, Turk A, Napiontek U, Keilmann A, Müller-Forell W, Kopp P, Pohlenz J. J Clin Endocrinol Metab. 2006 Jul;91(7):2678-81. Epub 2006 May 9.
- www.audiologiainfantile.com Articolo del Dr. Castiglione A.
- www.orpha.net
- www.xagena.it
- www.sin-italy.org
|








{kind=link}